HPC and Workflows
Overview
As a research scientist at Colorado State University, I develop computational workflows to investigate biological questions in developmental gene regulatory networks and host-vector dynamics in infectious diseases. My work focuses on implementing and optimizing bioinformatics pipelines that transform complex genomic datasets into biological insights.
I leverage High Performance Computing systems (CURC Alpine) to build production-ready workflows that are reproducible, scalable, and tailored to answer specific research questions. My computational expertise in Linux/Unix systems and open source software deployment enables me to bridge the gap between published methodologies and practical implementation.
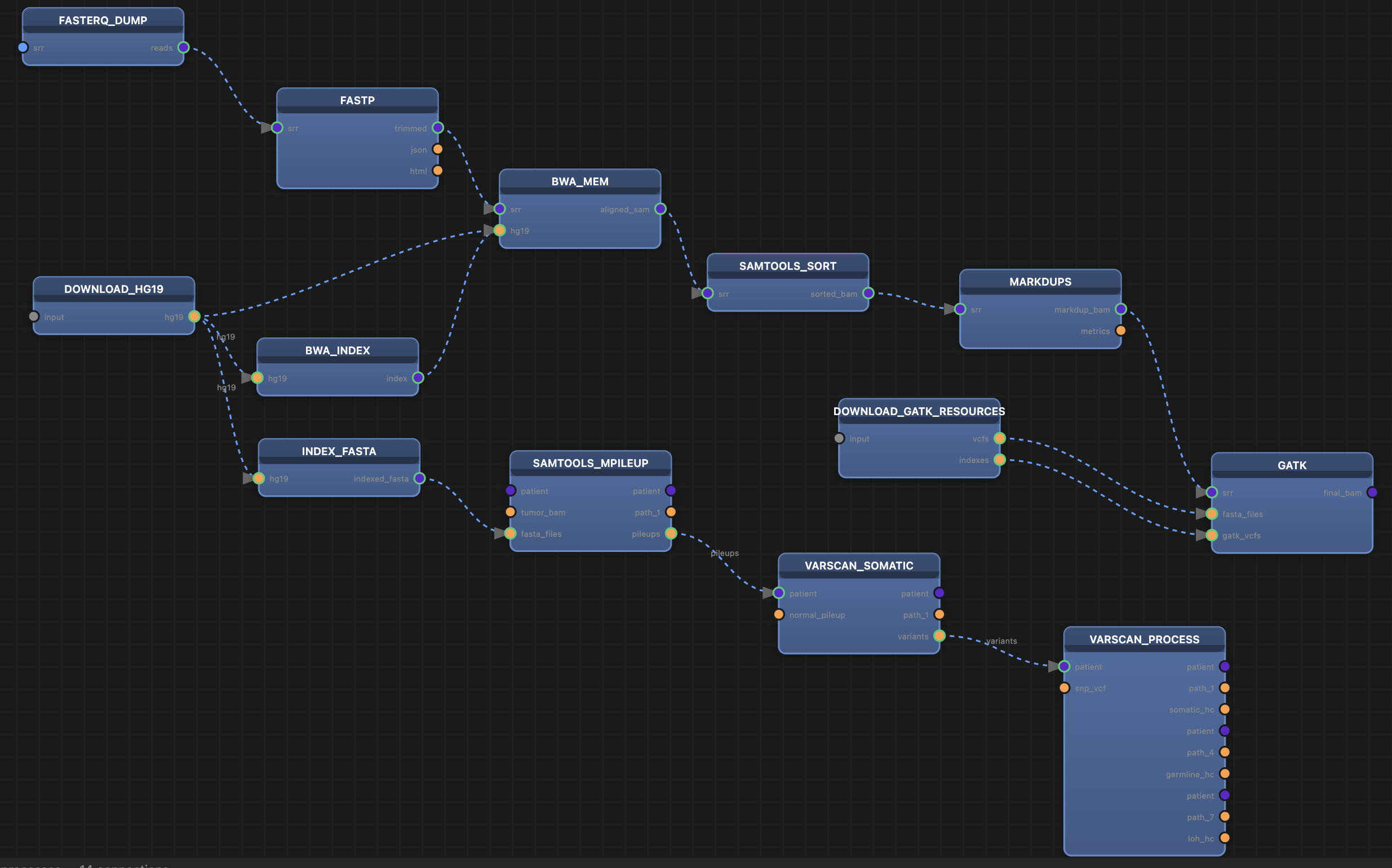
Research-Driven Workflow Development
Viral Variant Calling Pipeline
Nextflow | CSU/CURC Alpine
Adapted a complex Nextflow workflow for investigating viral evolution and host-vector interactions. Key contributions include:
- Designed filesystem organization strategy across /home, /project, and /scratch storage tiers for optimal performance on GPFS systems
- Optimized CPU, memory, and walltime requests to minimize queue wait times while maintaining throughput
- Modernized pipeline processes to improve code maintainability and execution efficiency
This deployment engineering work transformed a theoretical workflow into a production tool capable of processing large-scale viral genomic datasets.
modENCODE ChIP-seq Pipeline
SLURM/Bash | CSU/CURC Summit & Alpine
Implemented the complete modENCODE ChIP-seq analysis methodology for investigating transcription factor binding and gene regulation. Built a self-orchestrating SLURM pipeline featuring:
- Intelligent job management: Script detects execution context (launch vs. execution mode) and submits itself with proper SLURM dependencies (e.g.,
./pipeline.sh fastqc align bamgenerates chained jobs: fastqc → align → bam) - Complex branching logic: Handles pseudoreplicate-based quality control including parallel peak calling and correlation analysis for reproducibility assessment
- End-to-end processing: Raw sequencing reads through quality control, alignment, peak calling, and signal track generation
- Methodological contribution: Developed a pseudoreplicate seeding approach that was subsequently adopted by the ENCODE consortium
split-run-nf
Nextflow | Original Design
Created a Nextflow pipeline from scratch to parallelize bioinformatics tools that lack native multi-threading support. The workflow intelligently splits large FASTQ files into chunks, processes them concurrently across multiple nodes, and merges results—dramatically reducing wall-clock time for computationally intensive single-threaded tools.
Supporting Workflows
RNA-seq Analysis Pipelines
RNA-seq Array Processing Redesigned an RNA-seq workflow to eliminate directory collisions caused by Hisat2’s temporary files during parallel SLURM array execution. This refactoring enabled concurrent sample processing without interference, significantly reducing total processing time for large datasets.
Mouse Genome RNA-seq Pipeline Containerized end-to-end workflow (reads to gene counts) using Docker/Singularity. Ensures reproducible results across computing environments and simplifies dependency management.
SLURM Array Patterns Advanced SLURM job arrays demonstrating branching and convergence patterns using native scheduler commands for efficient resource utilization and dependency management.
HPC Implementation Expertise
Throughout my work at CSU, I’ve supported labs, courses, and collaborators in deploying bioinformatics workflows on HPC systems:
- Systems: CURC Summit, CURC Alpine, CSU Riviera
- Specialization: SLURM optimization, filesystem strategy, resource tuning
- Open source troubleshooting: Adapted published tools for HPC environments
Notable Technical Contributions
deepTools Bug Fix Fixed a critical bug preventing cluster diagnostic plots from rendering, contributing to this widely-used genomics visualization toolkit.
Adapted Research Tools
- HyperTRIBE: Adapted workflow for identifying RNA editing sites and modifications in mRNA transcripts
- Java Genomics Toolkit: Enhanced functionality of this genomic analysis toolkit (similar to bedtools and Picard)
Philosophy
Effective computational workflows are essential infrastructure for biological discovery. My approach emphasizes:
- Reproducibility: Comprehensive version control and clear dependency management
- Practicality: Workflows optimized for real-world HPC constraints (queue times, storage, resources)
- Transparency: Well-documented code enabling collaboration and knowledge transfer
- Biology-driven design: Computational solutions tailored to specific research questions
By building robust workflow infrastructure, I ensure that computational analysis serves as a reliable foundation for advancing biological understanding.