HPC and Workflows
Overview
As a research scientist at Colorado State University, I develop computational workflows to investigate biological questions in developmental gene regulatory networks and host-vector dynamics in infectious diseases. My work focuses on implementing and optimizing bioinformatics pipelines that transform complex genomic datasets into biological insights.
Research and Diagnostics Driven Workflow Development
Nextflow
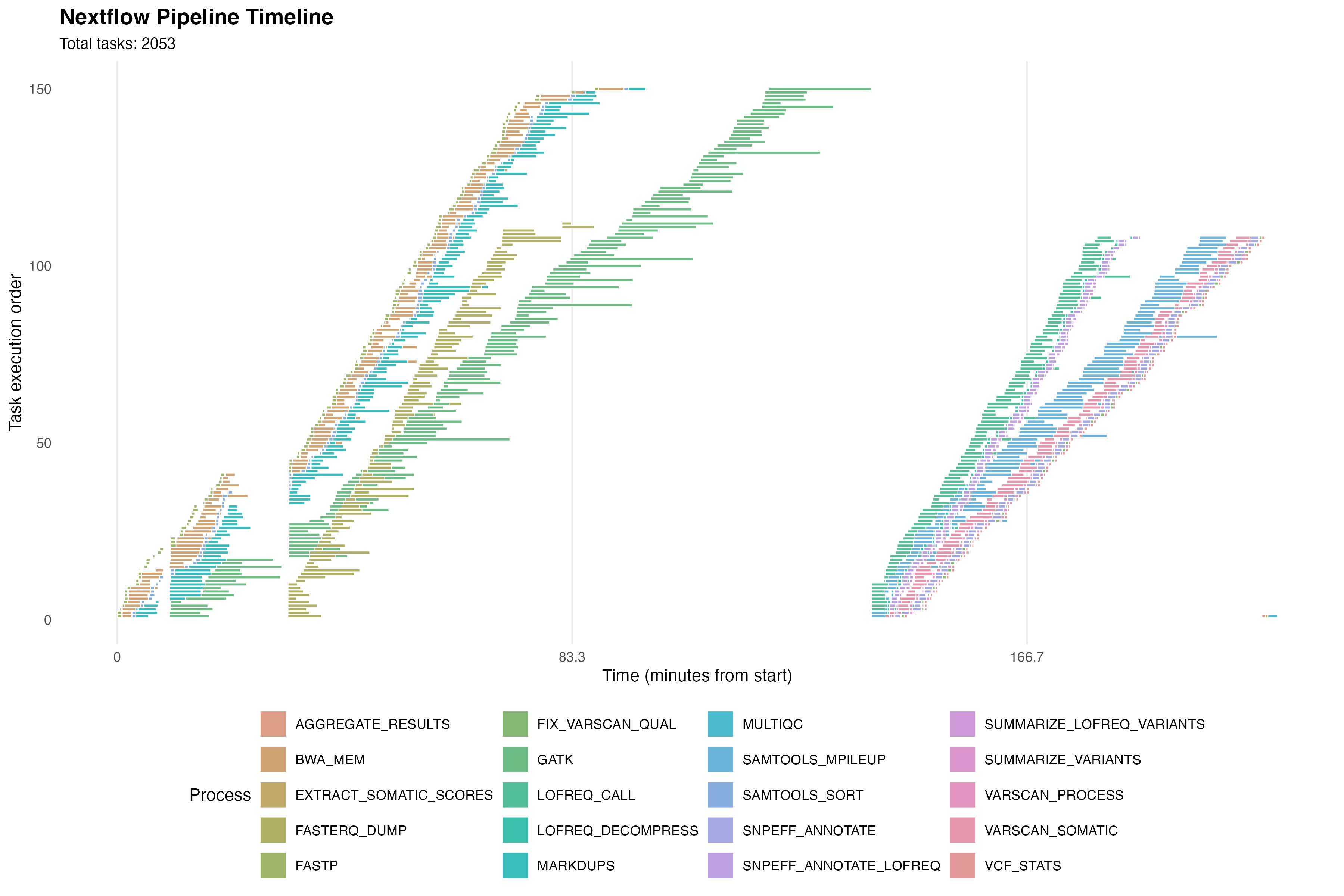
Somatic Tumor Variant Calling Pipeline
This nextflow pipeline calls variants on Illumina reads and has modifications to track mutations in tumor biopsies after Lim et al. (2021)1, which follows patients with colorectal cancer who become resistant to anti-EGFR treatment.
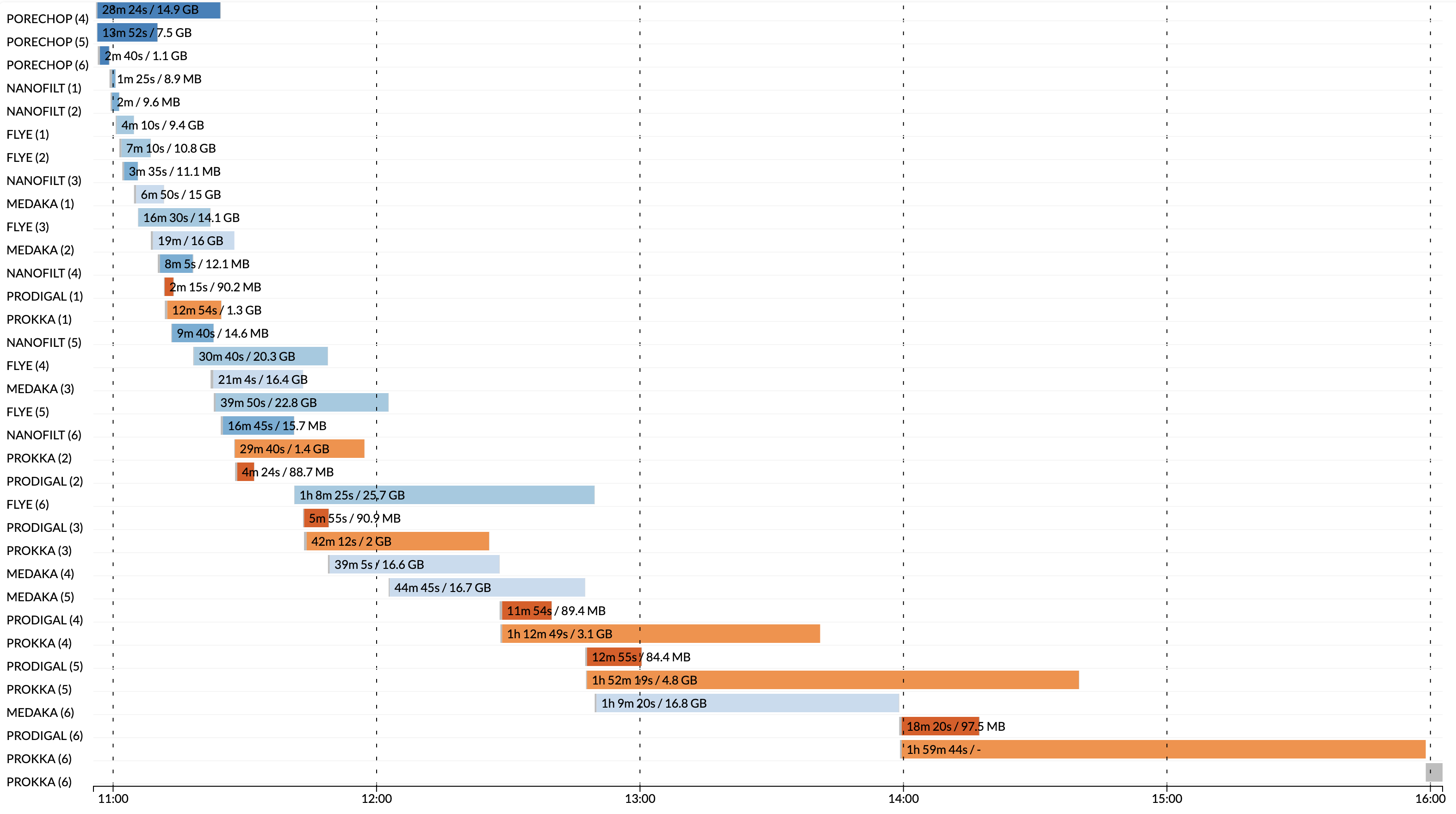
Long Read Assembly and Metagenomics Pipeline
This Nextflow pipeline is adapted from shell scripts for metagenomic analysis of long read sequencing of bacterial isolates.
SLURM
Submitter-Runner
Some pipelines are simple enough to implement with bash and SLURM dependency flags or arrays. The modENCODE Chip-seq analysis pipeline is not one of those, however- a workflow manager would have been perfect for the consortium. My submitter/runner hybrid mode script is a lite-weight substitute. It offers more control over job step invocation and runs without a parent control process (as needed by Nextflow).
ModENCODE Chip-seq analysis
We needed an in-house implementation of modENCODE’s ChIP-seq analysis methodology for investigating transcription factor binding and gene regulation. Our lab used this pipeline to reproduce the results coming out of the consortium (our study was later published in Williams et al, 2023).
It processes raw sequencing reads through quality control, alignment, peak calling, signal track generation, and statistical reproducibility analysis.

Sidenote: statistical reproducibility
Although my primary goal was to reproduce and pre-existing workflow, the developers adopted my approach for seeding pseudoreplicates. The prior method reused the same hard-coded value to seed subsampling procedures across all read files, potentially introducing bias.
Arrayed RNA-seq Analysis Pipelines
RNA-seq Array Processing Redesigned an RNA-seq workflow to eliminate directory collisions caused by Hisat2’s temporary files during parallel SLURM array execution. This refactoring enabled concurrent sample processing without interference, significantly reducing total processing time for large datasets.
Mouse Genome RNA-seq Pipeline Containerized end-to-end workflow (reads to gene counts) using Docker/Singularity. Ensures reproducible results across computing environments and simplifies dependency management.
SLURM Array Patterns Advanced SLURM job arrays demonstrating branching and convergence patterns using native scheduler commands for efficient resource utilization and dependency management.
HPC Implementation Expertise
Throughout my work at CSU, I’ve supported labs, courses, and collaborators in deploying bioinformatics workflows on HPC systems:
- Systems: CURC Summit, CURC Alpine, CSU Riviera
- Specialization: SLURM optimization, filesystem strategy, resource tuning
- Open source troubleshooting: Adapted published tools for HPC environments
Notable Technical Contributions
deepTools Bug Fix Fixed a critical bug preventing cluster diagnostic plots from rendering, contributing to this widely-used genomics visualization toolkit.
Adapted Research Tools
- HyperTRIBE: Adapted workflow for identifying RNA editing sites and modifications in mRNA transcripts
- Java Genomics Toolkit: Enhanced functionality of this genomic analysis toolkit (similar to bedtools and Picard)
Philosophy
Effective computational workflows are essential infrastructure for biological discovery. My approach emphasizes:
- Reproducibility: Comprehensive version control and clear dependency management
- Practicality: Workflows optimized for real-world HPC constraints (queue times, storage, resources)
- Transparency: Well-documented code enabling collaboration and knowledge transfer
- Biology-driven design: Computational solutions tailored to specific research questions
By building robust workflow infrastructure, I ensure that computational analysis serves as a reliable foundation for advancing biological understanding.
Footnotes and references
-
Circulating tumor DNA sequencing in colorectal cancer patients treated with first-line chemotherapy with anti-EGFR. Scientific Reports volume 11, Article number: 16333 (2021) ↩